What is Aicardi syndrome?
Aicardi syndrome is a rare neurological disorder. The severity of the syndrome and the associated signs and symptoms vary from person to person. The three main features of Aicardi syndrome are: Complete or partial absence of the nerve tissue that allows the right and left sides of the brain to communicate (corpus callosum)
What is the ICD-9 code for diagnosis?
ICD-9-CM 333.0 is a billable medical code that can be used to indicate a diagnosis on a reimbursement claim, however, 333.0 should only be used for claims with a date of service on or before September 30, 2015.
How many chromosomes does Aicardi syndrome have?
The few boys that have been identified with Aicardi syndrome have proved to have 47 chromosomes including an XXY sex chromosome complement, a condition called Klinefelter syndrome.
What is the ICD 9 code for Degen basal ganglia?
Short description: Degen basal ganglia NEC. ICD-9-CM 333.0 is a billable medical code that can be used to indicate a diagnosis on a reimbursement claim, however, 333.0 should only be used for claims with a date of service on or before September 30, 2015.
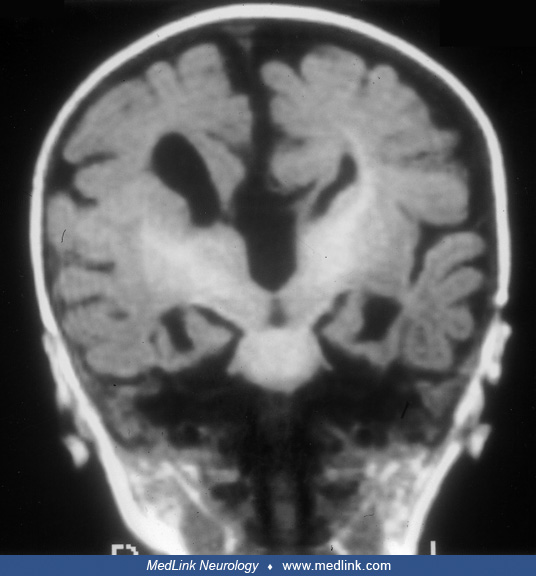
What is the ICD 10 code for Aicardi syndrome?
Congenital malformations of corpus callosum Q04. 0 is a billable/specific ICD-10-CM code that can be used to indicate a diagnosis for reimbursement purposes. The 2022 edition of ICD-10-CM Q04. 0 became effective on October 1, 2021.
What causes Aicardi Goutieres syndrome?
Aicardi-Goutieres syndrome-1 (AGS1) and AGS5 (an autosomal dominant form) are caused by a mutation in the TREX1 gene, AGS2 is caused by a mutation in the RNASEH2B gene, AGS3 is caused by a mutation in the RNASEH2C gene, AGS4 is caused by a mutation in the RNASEH2A gene.
How rare is Aicardi syndrome?
Aicardi syndrome is a very rare disorder. It occurs in about 1 in 105,000 to 167,000 newborns in the United States. Researchers estimate that there are approximately 4,000 affected individuals worldwide.
Is Aicardi-Goutières syndrome terminal?
As a result of the severe neurological problems usually associated with Aicardi-Goutières syndrome, most people with this disorder do not survive past childhood. However, some affected individuals who develop the condition later or have milder neurological problems live into adulthood.
Is Aicardi syndrome detectable prior to birth?
The diagnosis can be suspected by prenatal ultrasound with color Doppler identifying the agenesis of the corpus callosum. Usually, the diagnosis is confirmed in the neonate period by transfontanellar ultrasound and ophthalmological examination.
Is there a cure for Aicardi syndrome?
There is no cure for Aicardi syndrome nor is there a standard course of treatment. Treatment generally involves medical management of seizures and programs to help parents and children cope with developmental delays.
What are the symptoms of Aicardi syndrome?
What Are the Symptoms of Aicardi Syndrome?a coloboma, which is a hole or gap in one of the structures of the eye.abnormally small eyes.an unusually small head.hand deformities.intellectual disabilities.developmental delays.difficulty eating.diarrhea.More items...
When was Aicardi syndrome discovered?
Aicardi syndrome is a rare neurologic disorder first described by the French neurologist, Dr. Jean Aicardi, in 1965. It occurs almost exclusively in females (46,XX), however, it can also occur in males with Klinfelter Syndrome (47,XXY).
What are the defects of the brain that are common in Aicardi syndrome?
Other types of defects of the brain such as microcephaly, polymicrogyria, porencephalic cysts and enlarged cerebral ventricles due to hydrocephalus are also common in Aicardi syndrome.
How many chromosomes are in Aicardi syndrome?
Genetics. Almost all reported cases of Aicardi syndrome have been in girls. The few boys that have been identified with Aicardi syndrome have proved to have 47 chromosomes including an XXY sex chromosome complement, a condition called Klinefelter syndrome. All cases of Aicardi syndrome are thought to be due to new mutations.
What are the complications of aicardia?
Additional comorbidities and complications sometimes seen with Aicardi syndrome include porencephalic cysts and hydrocephalus, and gastro-intestinal problems. Treatment for porencephalic cysts and/or hydrocephalus is often via a shunt or endoscopic fenestration of the cysts, though some require no treatment.
How early can you get Aicardi syndrome?
Children are most commonly identified with Aicardi syndrome before the age of five months. A significant number of girls are products of normal births and seem to be developing normally until around the age of three months, when they begin to have infantile spasms. The onset of infantile spasms at this age is due to closure of the final neural synapses in the brain, a stage of normal brain development. A number of tumors have been reported in association with Aicardi syndrome: choroid plexus papilloma (the most common), medulloblastoma, gastric hyperplastic polyps, rectal polyps, soft palate benign teratoma, hepatoblastoma, parapharyngeal embryonal cell cancer, limb angiosarcoma and scalp lipoma.
Is Aicardi syndrome a mutation?
All cases of Aicardi syndrome are thought to be due to new mutations. No person with Aicardi syndrome is known to have transmitted the X-linked gene responsible for the syndrome to the next generation.
Is aicardi syndrome a developmental disability?
However, almost all people reported with Aicardi syndrome to date have experienced developmental delay of a significant degree, typically resulting in mild to moderate to profound intellectual disability. The age range of the individuals reported ...
When do children with AGS start having symptoms?
Children with later-onset AGS begin having symptoms after the first weeks or monhs or normal development. Then, they may experience:
What is AGS in medical terms?
Aicardi-Goutieres Syndrome (AGS) is a rare genetic neurodevelopmental disorder characterized by encephalopathy (brain dysfunction) that affects newborn infants and usually results in mental and physical disability.
What is the ICD code for corpus callosum?
Q04.0 is a billable ICD code used to specify a diagnosis of congenital malformations of corpus callosum. A 'billable code' is detailed enough to be used to specify a medical diagnosis.
Is a diagnosis present at time of inpatient admission?
Diagnosis was present at time of inpatient admission. Yes. N. Diagnosis was not present at time of inpatient admission. No. U. Documentation insufficient to determine if the condition was present at the time of inpatient admission. No.

Overview
Aicardi syndrome is a rare genetic malformation syndrome characterized by the partial or complete absence of a key structure in the brain called the corpus callosum, the presence of retinal abnormalities, and seizures in the form of infantile spasms.
Aicardi syndrome is theorized to be caused by a defect on the X chromosome as it has thus far only been observed in girls or in boys with Klinefelter syndrome. Confirmation of this theory awai…
Signs and symptoms
Children are most commonly identified with Aicardi syndrome before the age of five months. A significant number of girls are products of normal births and seem to be developing normally until around the age of three months, when they begin to have infantile spasms. The onset of infantile spasms at this age is due to closure of the final neural synapses in the brain, a stage of normal brain development . A number of tumors have been reported in association with Aicardi syndrome: choroid …
Genetics
Almost all reported cases of Aicardi syndrome have been in girls. The few boys that have been identified with Aicardi syndrome have proved to have 47 chromosomes including an XXY sex chromosome complement, a condition called Klinefelter syndrome.
All cases of Aicardi syndrome are thought to be due to new mutations. No person with Aicardi syndrome is known to have transmitted the X-linked gene responsible for the syndrome to the n…
Diagnosis
Aicardi syndrome is typically characterized by the following triad of features - however, one of the "classic" features being missing does not preclude a diagnosis of Aicardi Syndrome, if other supporting features are present.
1. Partial or complete absence of the corpus callosum in the brain (agenesis of the corpus callosum);
Treatment
Treatment of Aicardi syndrome primarily involves management of seizures and early/continuing intervention programs for developmental delays. Additional comorbidities and complications sometimes seen with Aicardi syndrome include porencephalic cysts and hydrocephalus, and gastro-intestinal problems. Treatment for porencephalic cysts and/or hydrocephalus is often via a shunt or endoscopic fenestration of the cysts, though some require no treatment. Placement of a f…
Prognosis
The prognosis varies widely from case to case, depending on the severity of the symptoms. However, almost all people reported with Aicardi syndrome to date have experienced developmental delay of a significant degree, typically resulting in mild to moderate to profound intellectual disability. The age range of the individuals reported with Aicardi syndrome is from birth to the mid-40s. There is no cure for this syndrome.
Epidemiology
Worldwide prevalence of Aicardi syndrome is estimated at several thousand, with approximately 900 cases reported in the United States.
History
This disorder was first recognized as a distinct syndrome in 1965 by Jean Aicardi, a French pediatric neurologist and epileptologist.
Popular Posts:
- 1. icd 10 code for skin rash unspecified
- 2. icd-10-pcs code for insertion of hemodialysis catheter
- 3. icd 10 code for iup unspecified
- 4. icd 10 code for follow up of blood sugar readings
- 5. icd 10 code for atypical depressive disorder (icd-9)
- 6. icd 10 code for acute organ dysfunction
- 7. what is the correct icd 10 code for group a streptococcus
- 8. icd 10 code for gsw to buttocks
- 9. icd 10 code for malfunction of suprapubic catheter
- 10. icd-10 code for irritable bowel syndrome